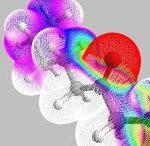
 The picture shows the electrostatic field of a d-fructose molecule which must be reproduced in an efficient manner in a molecular dynamics simulation. Electrostatic forces dominate the long-range interactions between molecules and must be accounted for in atomistic simulations of large systems. Since these forces are required at each step in a molecular dynamics or Monte Carlo simulation, it is important to develop efficient methods that accurately reproduce the electrostatic fields of molecules. Traditionally, charge-equilibration, or fluctuating-charge techniques have been used to generate the electrostatic fields of molecules, yet their application remains limited due to their inaccuracy. We have developed a Split-Charge Equilibration technique, in which bond-dependent parameters are introduced into the model. The approach is more accurate than previous methods, and can be extended to address nonlocalized bond making/breaking events (Chemistry!) in large systems.
The picture shows the electrostatic field of a d-fructose molecule which must be reproduced in an efficient manner in a molecular dynamics simulation. Electrostatic forces dominate the long-range interactions between molecules and must be accounted for in atomistic simulations of large systems. Since these forces are required at each step in a molecular dynamics or Monte Carlo simulation, it is important to develop efficient methods that accurately reproduce the electrostatic fields of molecules. Traditionally, charge-equilibration, or fluctuating-charge techniques have been used to generate the electrostatic fields of molecules, yet their application remains limited due to their inaccuracy. We have developed a Split-Charge Equilibration technique, in which bond-dependent parameters are introduced into the model. The approach is more accurate than previous methods, and can be extended to address nonlocalized bond making/breaking events (Chemistry!) in large systems.